Main scientific focus areas:
To investigate how deregulation of the Writers, Erasers and Readers of histone methylation, focused on histone H3 lysine 36 methylation (H3K36me), promote tumorigenesis, with a focus on lung and pancreatic cancers
To uncover new roles for non-histone methylation in the regulation of fundamental processes and understand connections to human diseas
By analogy to the regulation of transcription by histone methylation, we are exploring roles for protein methylation in shaping the proteome through regulation of mRNA translation elongation. We have identified KMTs devoted to modifying the translation machinery that promote tumorigenesis. Current work aims to understand the mechanisms of action for these enzymes in solid tumors and to evaluate their suitability as clinical targets.
Beyond the ~25 KMTs involved in histone methylation, there are many other ‘orphan’ or candidate KMTs. We have established a robust workflow for uncovering the catalytic, biological and pathologic functions of these enzymes
Identify Readers of non-histone methylation
We recently identified the first known metazoan protein histidine methyltransferase. Mass spectrometry studies suggest that there are hundreds of histidine methylated proteins in the human proteome. We are interested in understanding the broad role for histidine methylation in the regulation of protein behavior and signal transduction, with a focus on chromatin biology and gene expression, through the study of the Writers, Readers, and potentially Erasers for this previously little characterized post-translational modification.
To develop proteomic and chemical methods, tools, reagents, and technologies to study the biology and pathology of protein methylation
To perform proof of concept pre-clinical studies with inhibitors of KMTs to identify suitable indications to guide clinical studies
Lysine methylation biology
Background:
Covalent post-translational modifications (PTMs) of proteins contribute to all aspects of cell physiology and are a major source of molecular functional diversity in mammalian cells. Aberrant regulation of PTMs is a common feature of human diseases like cancer. A highly regulated set of enzymes catalyzes the addition and removal of PTMs, often in response to diverse signals. For example, the lysine methylation chemical reaction is the addition of one, two or three methyl groups to the ε-nitrogen of a lysine sidechain, forming mono-, di-, and tri-methylated derivatives (see Figure below). This reaction, while subtly changing the primary structure of the modified polypeptide, greatly increases the information encoded within the molecule, highlighted by unique activities often coupled to the specific extent of methylation. The methylation of lysines on histone and non-histone proteins is catalyzed by protein lysine methyltransferases (KMTs) and removed by protein lysine demethylases (KDMs). In the human genome, there are predicted to be well over 100 KMTs and mass spectrometry-based studies suggest the existence of 1000s of lysine methylation events across the human proteome.
Lysine methylation reaction: The chemical structure of lysine and methylated derivatives. Lysine methylation is performed KMTs, which replace the hydrogen moiety on the side chain with one, two or three methyl groups. Methyl groups can be removed by KDMs.
Histone Lysine Methylation
A highly complex molecular network at chromatin regulates eukaryotes genomes, with all DNA-templated processes being fundamentally affected by chromatin structure and dynamics. One of the major mechanisms for chromatin regulation involves the reversible covalent post-translational modification of histone proteins by chemical moieties such as acetyl-, methyl- and phospho- groups. These different histone modifications are linked to discrete chromatin states and are thought to regulate the extent of accessibility of DNA to transacting factors. Of the various histone modification systems, histone methylation is the most diverse with respect to the number of residues targeted for modification, potential for signaling, and biological functions regulated.
There are a large number of enzymes that catalyze the addition (often referred to as “writers”) or removal (referred to as “erasers”) of specific histone methylation events. At the molecular level, the addition of a methyl moiety to a protein serves as a signal to directly regulate modular protein-protein interactions. In this context, the proteins and domains that recognize distinct histone methylation events, often referred to as “readers” or “effectors”, are postulated to define the functional consequences of specific modifications by transducing molecular events at chromatin to distinct biological outcomes. Indeed, histone methylation has been clearly linked through specific reader proteins, including several identified by our group and collaborators, to fundamental DNA-templated processes, including transcriptional activation and repression [PDF], DNA repair, DNA recombination [PDF], DNA replication [PDF], and chromosome segregation. The methylation of histones has also been shown to be a critical mechanism employed for the regulation of epigenetic processes, and deregulation in histone methylation dynamics is clearly implicated in human diseases ranging from cancer and aging to disorders of developmental and cognitive function.
NSD enzymes, H3K36 Methylation and Cancer
Methylation at H3K36 is a key epigenetic modification. Mutations in the different H3K36 KMTs are linked to a variety of cancer types and developmental disorders. SETD2, which synthesizes H3K36me3 in humans, regulates DNA methylation, RNA processing and DNA repair. SETD2 is a tumor suppressor and commonly mutated in broad types of cancer. H3K36me2 is linked to DNA methylation, gene activation, and cellular transformation. There are four enzymes that generate H3K36me2: NSD1, NSD2, NSD3, and ASH1L. In contrast to SETD2, the H3K36me2-specific KMTs promote oncogenesis due to genetic alterations such as gene amplifications, translocations, gain-of-function mutations, and over-expression.
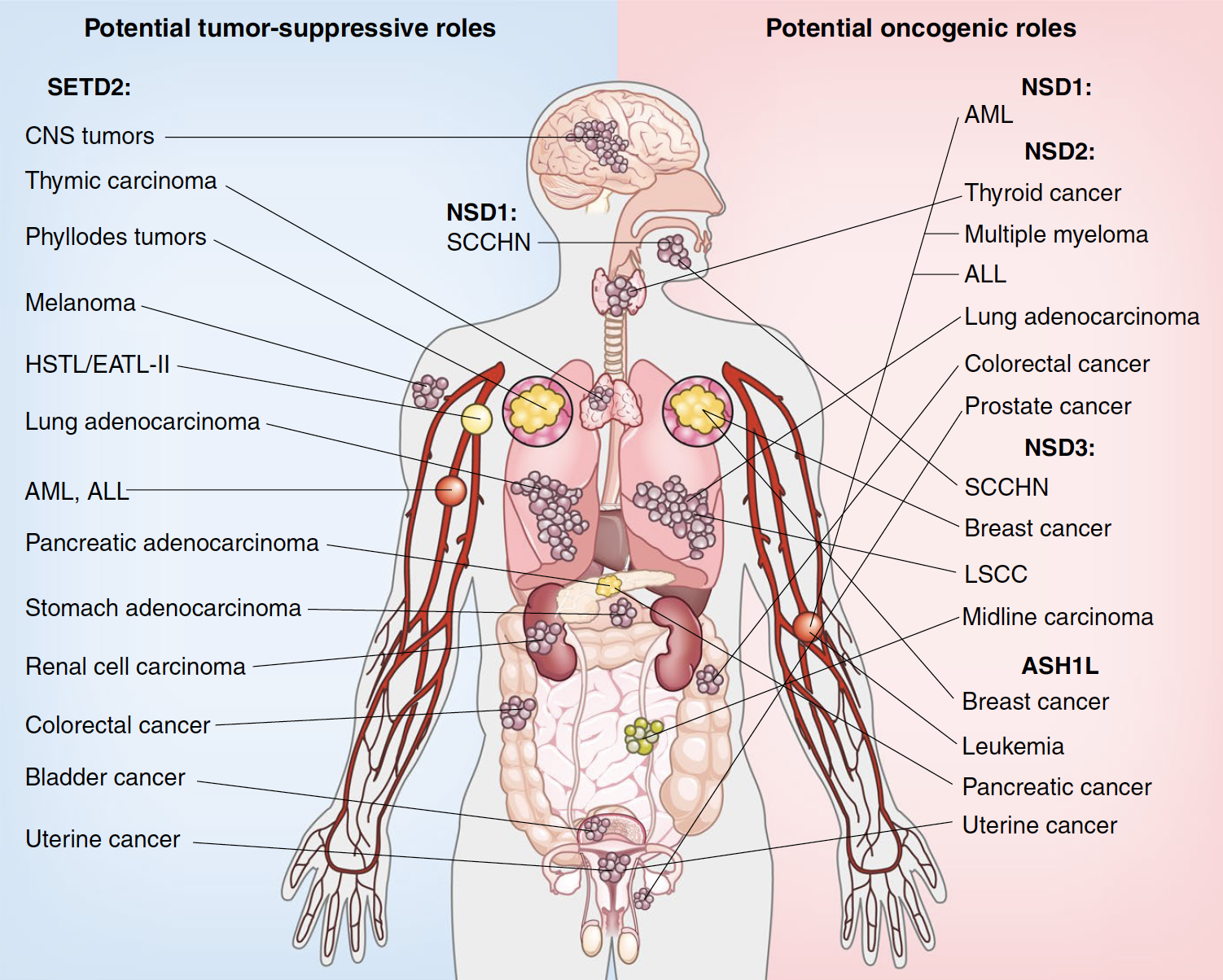
Examples of cancers associated with H3K36 methyltransferases
We have several ongoing projects exploring the mechanism of action for the different H3K36 methylating enzymes in the regulation of gene expression, chromatin, and epigenetics and how these processes influence oncogenesis in different cancer contexts.
Non-histone protein lysine methylation
Over the last decade, there is a growing appreciation in the field that a number of non-histone proteins, including several with clear roles in transcription, signal transduction, and cancer biology are lysine methylated (e.g. p53 [PDF] and RelA [PDF]). There are also several enzymes devoted to methylating factors involved in protein synthesis, particularly the translation elongation step. Indeed, there are >200 proteins in the human genome that harbor either a SET or seven-b-strand (7bS) catalytic methylation domain, and an increasing number of examples of mutation or translocation of known or candidate KMT genes being linked to human disorders. Thus, it is likely that deregulation in non-histone protein methylation homeostasis plays a crucial role in disease pathogenesis. However, despite the fundamental role for lysine methylation in biology and disease, the catalytic activity, substrate specificity, and biological function for many potential KMT enzymes is not known, and possible connections between non-histone methylation and human health and disease is vastly underexplored.
Over the last several years we have used several different strategies to characterize and identify physiologic substrates for many KMTs, including: (i) biochemical candidate approaches, (ii) protein arrays, (iii) chemical biology, (iv) lysine methylomics, (v) KMT-focused CRISPR-based biochemical and mass spectrometry screening and (vi) mass spectrometry coupled to activity-based biochemical fractionation in genetically modified lysates.
Current projects focus on several orphan enzymes that are implicated in solid tumor etiology and in elucidating signaling functions for lysine methylation in the regulation of protein synthesis, with a focus on mRNA translation elongation
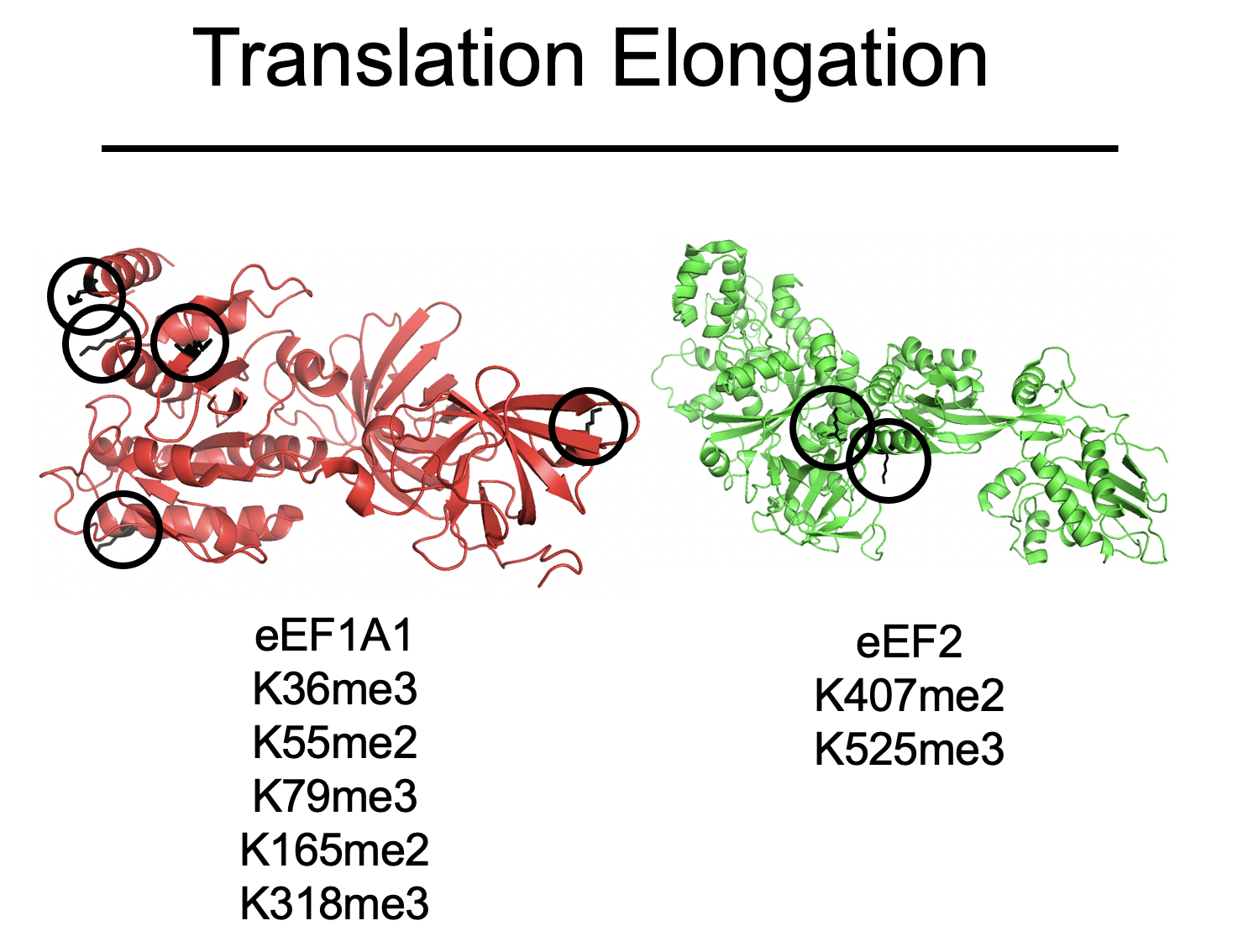
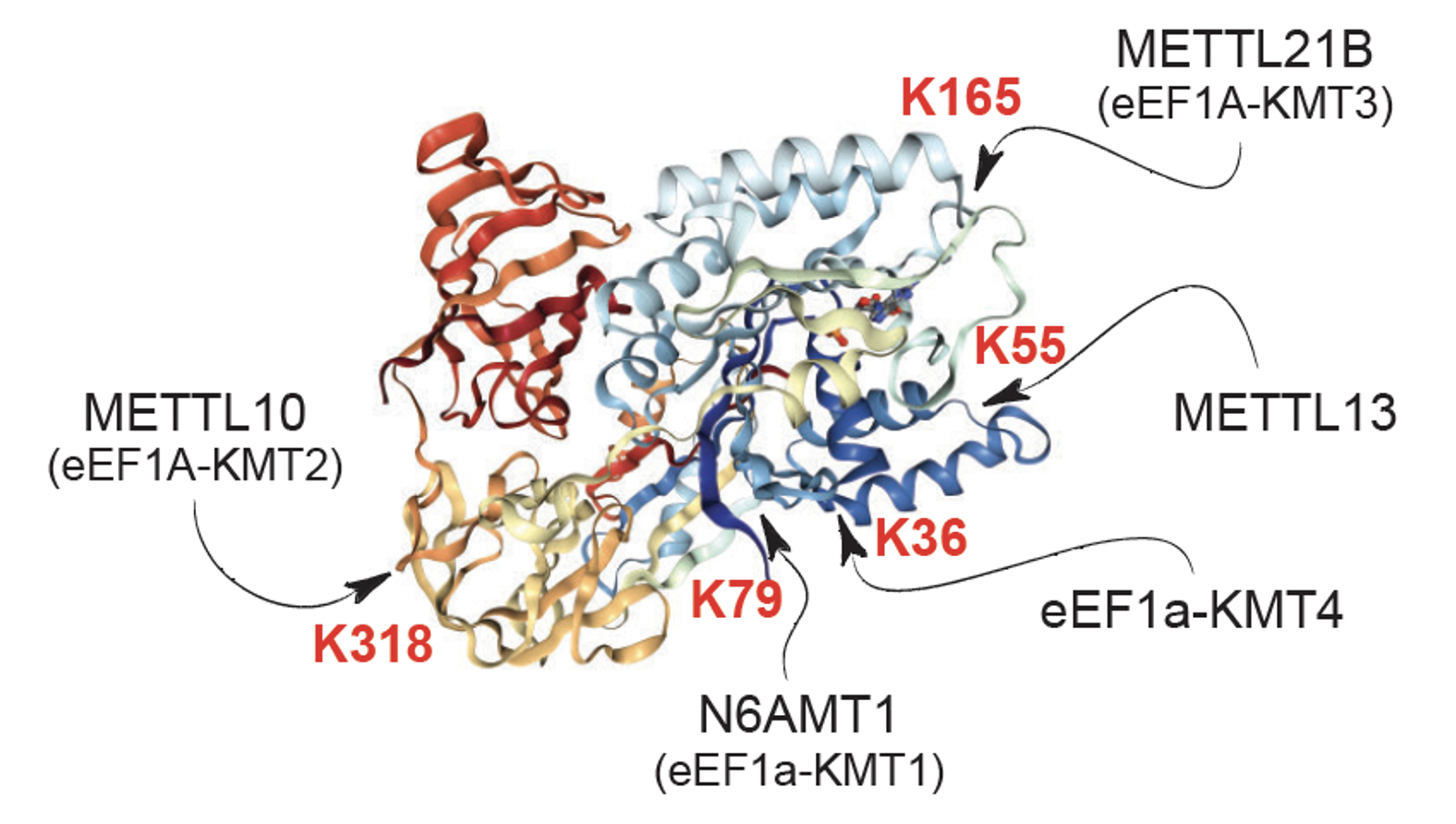
Top: Sites of methylation on eEF1A and eEF2
Bottom: eEF1A KMTs and their substrate sites
Want to learn more - some Reviews from the lab:
Bhat KP, Umit Kaniskan, Jin J and O Gozani. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. 2021. Nature Review Drug Discovery [PDF]
D Husmann and O Gozani. Histone lysine methyltransferases in biology and disease. 2019. Nature Structure and Molecular Biology [PDF]
SM Carlson and O Gozani. Nonhistone lysine methylation in the regulation of cancer pathways. 2016. Cold Spring Harbor Perspectives Medicine [PDF]
van Nuland R and O Gozani. Histone H4 Lysine 20 (H4K20) methylation, expanding the signaling potential of the proteome one methyl moiety at a time. 2016. Molecular Cell Proteomics. [PDF]
Carlson SM and O Gozani. Emerging technologies to map the protein methylome. 2014. Journal of Molecular Biology. [PDF]
Moore KE and O Gozani. An unexpected journey: lysine methylation across the proteome. 2014. Biochim Biophysics Acta. [PDF]
Wilkinson AW and O Gozani. Histone-binding domains: strategies for discovery and characterization. 2014. Biochim Biophysics Acta. [PDF]
West LE and O Gozani. Regulation of p53 function by lysine methylation. 2011. Epigenomics [PDF]
Shi X and O Gozani. The fellowships of the INGs. 2005. J Cell Biochemistry. [PDF]
Some of our recent publications:
Yuan et al., Nature 2021: Elevated NSD3 histone methylation activity drives squamous cell lung cancer [PDF]
Lung squamous cell carcinoma (LUSC) is a leading cause of cancer-related mortality worldwide. This malignancy is characterized by well-defined driver mutations, with 20% of patients having FGFR1 gene amplification. However, the causative nature of FGFR1 amplification in LUSC was unclear as clinical trials with FGFR1 inhibitors as a targeted therapy have been unsuccessful. The FGFR1 gene co-amplifies with other neighboring genes in the 8p11-12 genomic region, a major amplicon associated with multiple cancers including LUSC, raising the possibility of a different gene as the as the major mutational driver in the 8p11-12 amplicon in LUSC.In collaboration with the Mazur lab and several other groups, we found that in LUSC patients, NSD3 is the most frequently amplified gene in the 8p11-12 genomic region. Moreover, the expression of NSD3, but not of FGFR1, significantly correlated with gene amplification, and more generally, NSD3 is overexpressed in 60% of LUSC patients. We developed a robust mouse model of LUSC and found that deletion of NSD3, but not FGFR1, attenuated tumorigenesis and prolongs survival in vivo. By screening cancer-associated mutations, we identified a recurrent gain-of-function NSD3 variant (NSD3T1232A) with enhanced H3K36me2 catalytic activity. NMR-based structural dynamics experiments revealed that the T1232A mutation relieves an auto-inhibitory conformation and conferred active-site accessibility to increase NSD3’s enzymatic activity. We leveraged NSD3T1232A hyperactivity to model NSD3 amplification in vivo in LUSC mouse models. These studies showed that transgenic expression of NSD3T1232A and increased H3K36me2 have the opposite effect as the NSD3 knockout, accelerating tumorigenesis and lethality in vivo. Integrative RNA-seq and CUT&RUN-based chromatin profiling analyses in LUSC-derived cell lines demonstrated that NSD3, via H3K36me2, drives an oncogenic gene expression program that includes stimulation of targets involved in mTOR signaling and MYC-associated pathways. We also found that NSD3’s catalytic activity promotes (1) oncogenic transformation of human tracheobronchial cell and (2) xenograft growth of multiple human 8p11-12-amplified LUSC cell lines. Further, NSD3 deletion of NSD3 and reduction of H3K36me2 inhibited tumor growth in patient-derived xenografts (PDXs) from primary human LUSC tumors. Finally, we found that the oncogenic advantage provided by NSD3 to LUSC tumors comes with the cost of hypersensitivity to bromodomain inhibition (BETi), a clinically actionable vulnerability.Thus, this study uncovered a crucial role for NSD3 in LUSC pathogenesis and provides a potential epigenetic-based clinical path forward for treating 8p11-12-positive LUSC patients. Further, our work has broad implications for NSD3-mediated chromatin regulation in oncology, as in addition to LUSC, the 8p11-12 amplicon is a common molecular signature of breast cancer and many other malignancies.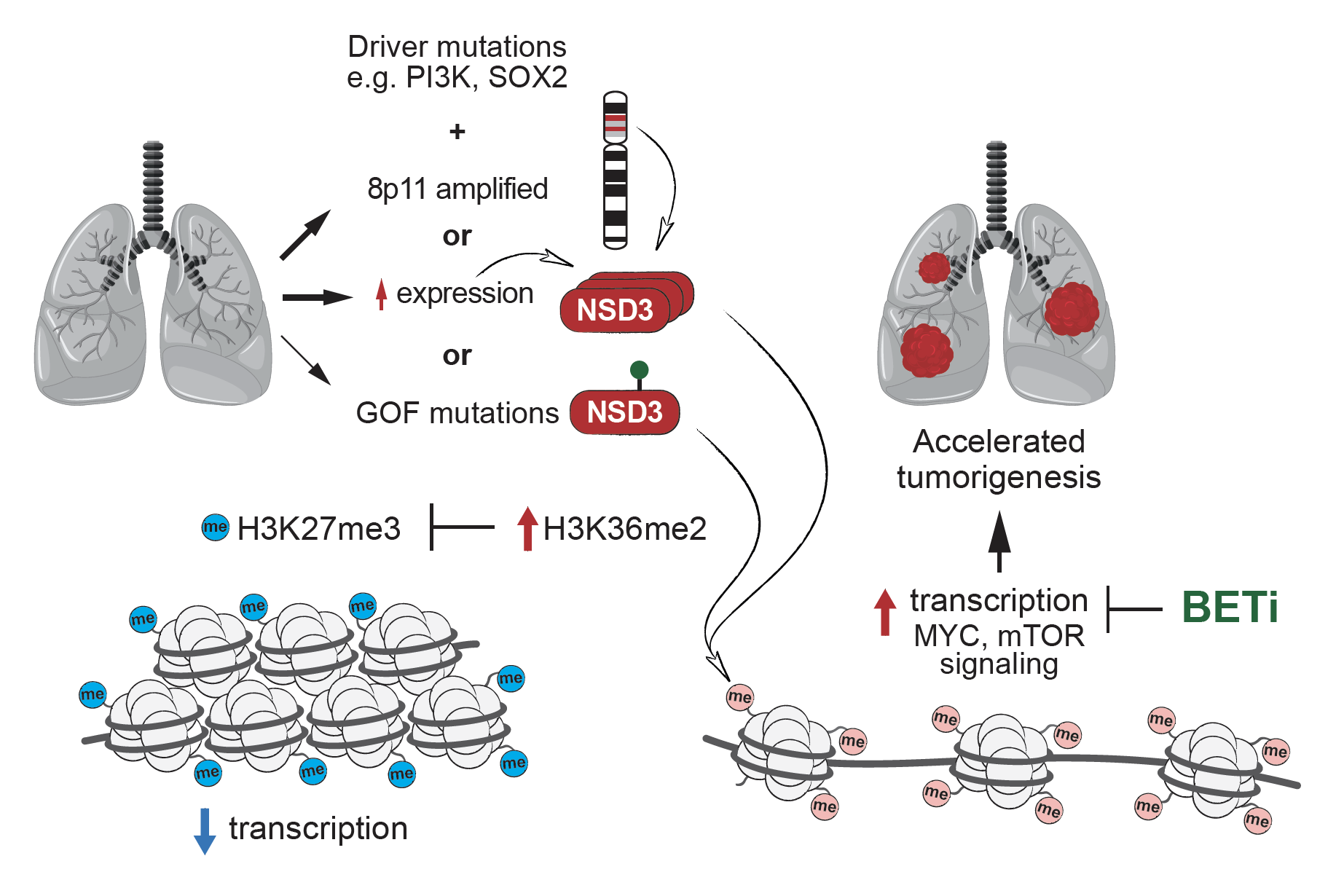
Model for how increased NSD3 H3K36me2 activity promotes lung squamous cell cancer
Wang et al., Cancer Cell 2020: SETD5-Coordinated Chromatin Reprogramming Regulates Adaptive Resistance to Targeted Pancreatic Cancer Therapy [PDF]
In collaboration with the Mazur lab and others, we identified SETD5 as a clinically actionable master epigenetic regulator of adaptive targeted therapy resistance in pancreatic ductal adenocarcinoma (PDAC), a deadly cancer with no effective treatments. Using patient derived PDAC xenografts and an innovative dual-recombinase mouse model that allows for acute deletion of Setd5 in tumors in vivo, we found that SETD5 deletion in malignant PDAC refractory to MEK inhibition (MEKi) restores the vulnerability of these tumors to targeted MEKi therapy. We also demonstrated that SETD5 was incorrectly characterized as being an active enzyme and rather has a novel biochemical scaffolding function within a distinct co-repressor complex that selectively swaps the activating acetylation mark with repressive methylation at lysine 9 of histone H3 (H3K9) at gene targets. Through these activities, the SETD5 complex regulates expression of an extensive gene network of known drug resistance pathways to reprogram cellular responses to MEKi. Finally, we showed that pharmacologic blockade with small molecule inhibitors of the histone-modifying activities associated with the SETD5 complex disrupt the SETD5-driven resistance program and sustain inhibition of tumor growth by MEKi in pre-clinical murine and human models of PDAC.
This work uncovered, to our knowledge, the first known chromatin-based mechanisms that is specifically induced to mediate adaptive resistance to targeted therapy, identified a drug resistance pathway that is clinically actionable and of high therapeutic relevance for PDAC and several other cancer types, and revived a clinical path for rationale deployment of FDA-approved MEK inhibitors to treat PDAC, a lethal cancer with at present limited treatment options.
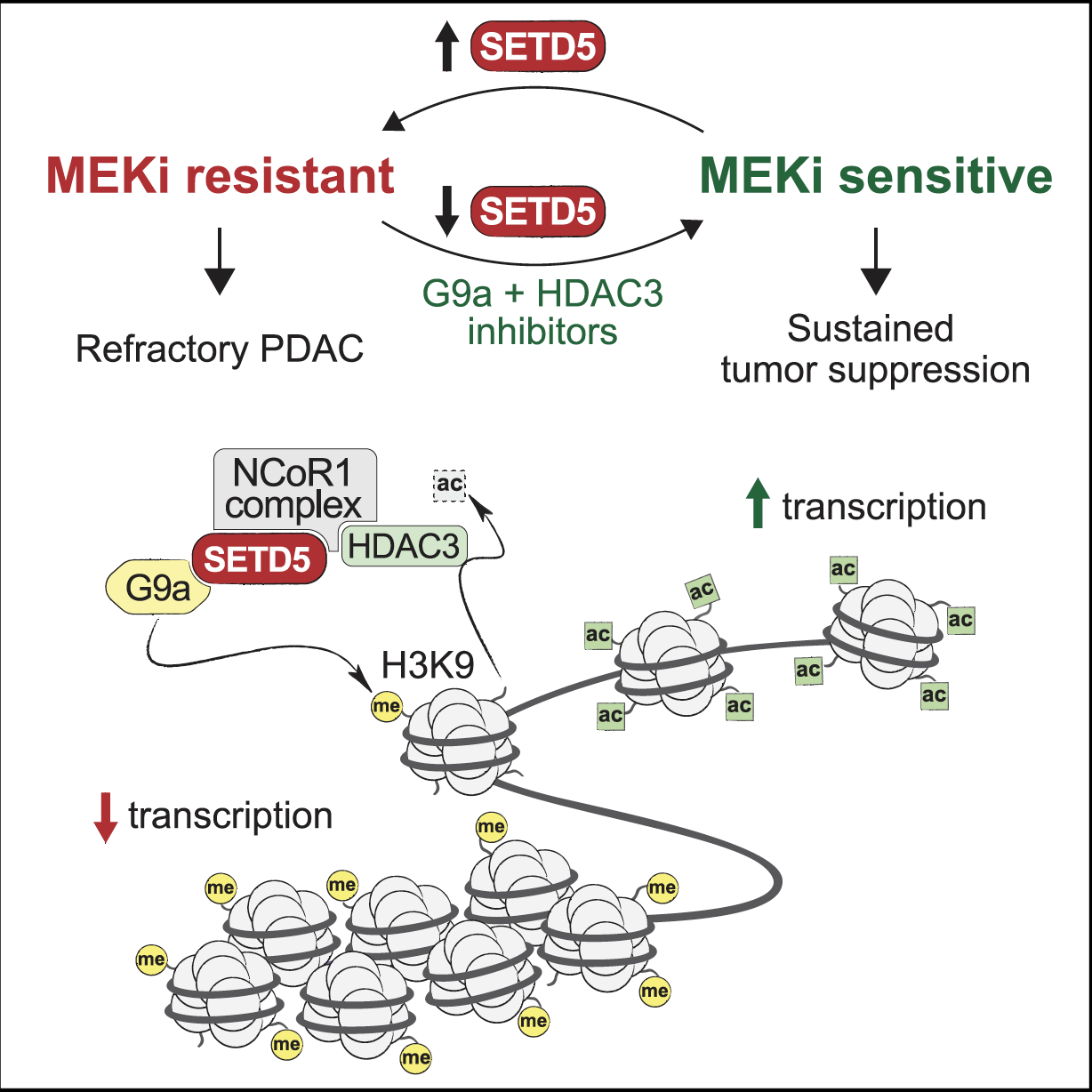
Model for SETD5-mediated drug resistance programming at chromatin in pancreatic cancer
Liu et al, Cell 2019: METTL13 Methylation of eEF1A Increases Translational Output to Promote Tumorigenesis [PDF]
Mechanisms to increase protein synthesis are crucial in the development and progression of diverse cancers. While most oncogenic pathways target the translation initiation machinery, the elongation step of translation also represents an important regulatory node. We found that METTL13 (methyltransferase like 13) di-methylation of eEF1A (eukaryotic elongation factor 1A) at lysine 55 (eEF1AK55me2) is an adaptation utilized by Ras-driven cancers to increase translational output and promote tumorigenesis in vivo. METTL13-catalyzed eEF1A methylation increased the intrinsic GTPase activity of eEF1A in vitro and protein production in cells. METTL13 expression and eEF1AK55me2 levels are specifically upregulated in cancer and negatively correlate with pancreatic and lung cancer patient survival. METTL13 deletion and subsequent loss of eEF1AK55me2 did not affect non-transformed cells but greatly attenuated Ras-driven neoplastic growth in mouse models and in patient-derived xenografts (PDX) from primary pancreatic and lung tumors. Finally, METTL13 depletion rendered cancer cells and PDX tumors highly susceptible to drugs that target growth pathways. These findings uncovered a mechanism by which lethal cancers become dependent on the METTL13-eEF1AK55me2 axis to meet their elevated protein synthesis requirement and suggest that METTL13 inhibition may constitute a targetable vulnerability of tumors driven by aberrant Ras-signaling.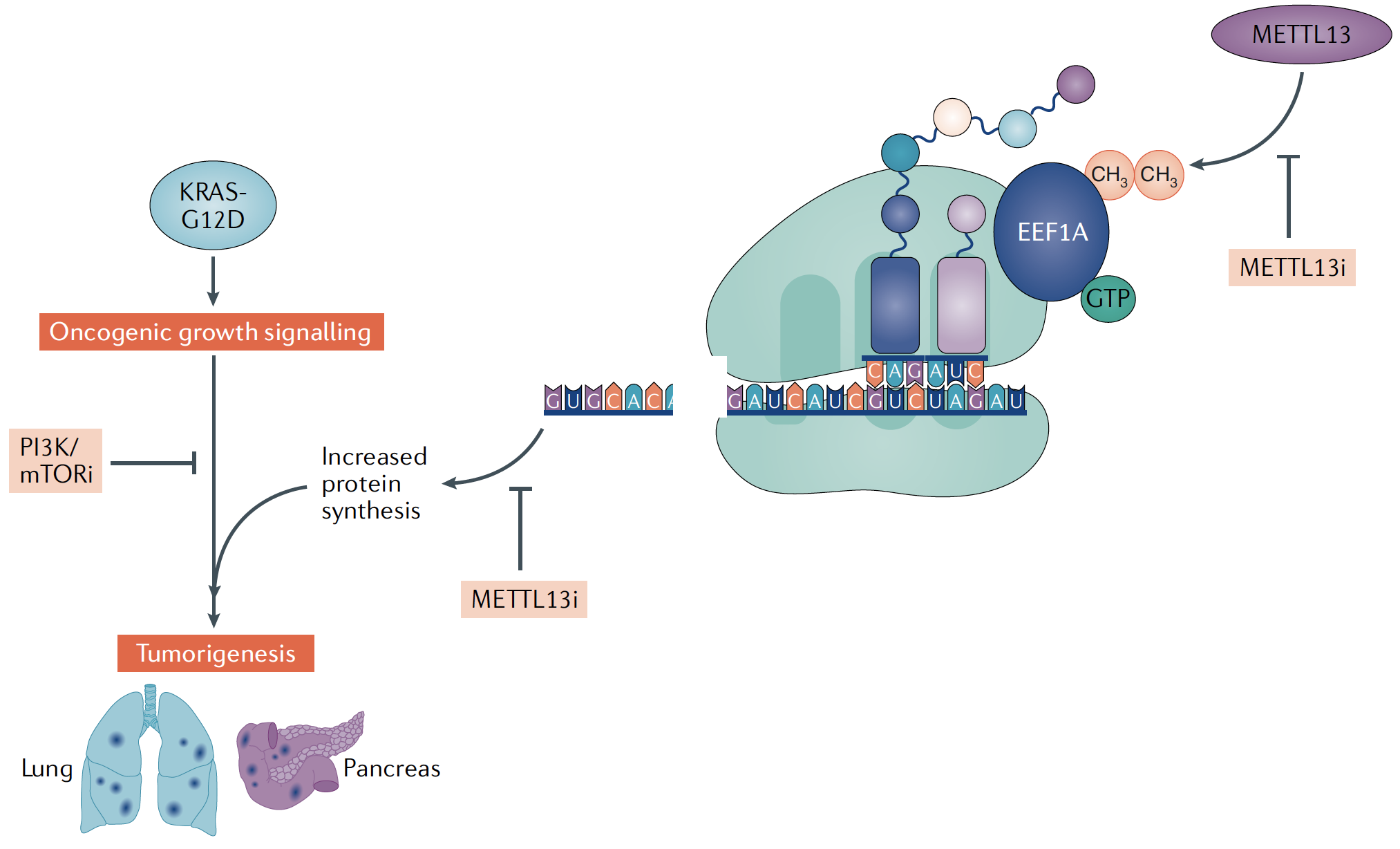
Model for METTL13 regulation of protein synthesis to promote KRAS-driven cancers
Wilkinson et al., Nature 2018: SETD3 is an actin histidine methyltransferase that prevents primary dystocia [PDF]
For over fifty years, the methylation of mammalian actin at histidine 73 (actin-H73me) has been known to exist. Beyond mammals, actin-H73me is not found in S. cerevisiae, but we found actin-H73 conserved in several other model animal and plant organisms (e.g. worms, flies, and Arabidopsis). Despite the pervasiveness of H73me, its function was enigmatic, and the enzyme generating this modification was unknown. We identified SETD3 (SETdomain protein 3) as the physiologic actin histidine 73 methyltransferase. SETD3 catalyzes histidine methylation on all actin isoforms in vitro and H73me reduces the nucleotide exchange rate on actin monomers to modestly accelerate actin filament assembly. SETD3 deficient mice showed complete loss of actin-H73me in multiple tissues and quantitative proteomics singled out actin-H73 as the principal physiologic SETD3 substrate. SETD3 deficient female mice have severely decreased litter sizes due to primary maternal dystocia that is refractory to ecbolic induction agents. Further, depletion of SETD3 impaired signal-induced contraction in primary human uterine smooth muscle cells. Together, our results identified the first mammalian protein histidine methyltransferase and uncovered a pivotal role for SETD3 and actin-H73me in the regulation of smooth muscle contractility. Further, SETD3 represents the first SET domain family member to methylate a residue besides lysine, broadening the current framework of possible chemistries catalyzed by SET domain enzymes. These and other data identifying many histidine methylated proteins suggest a broad role for protein histidine methylation in the regulation of mammalian proteomes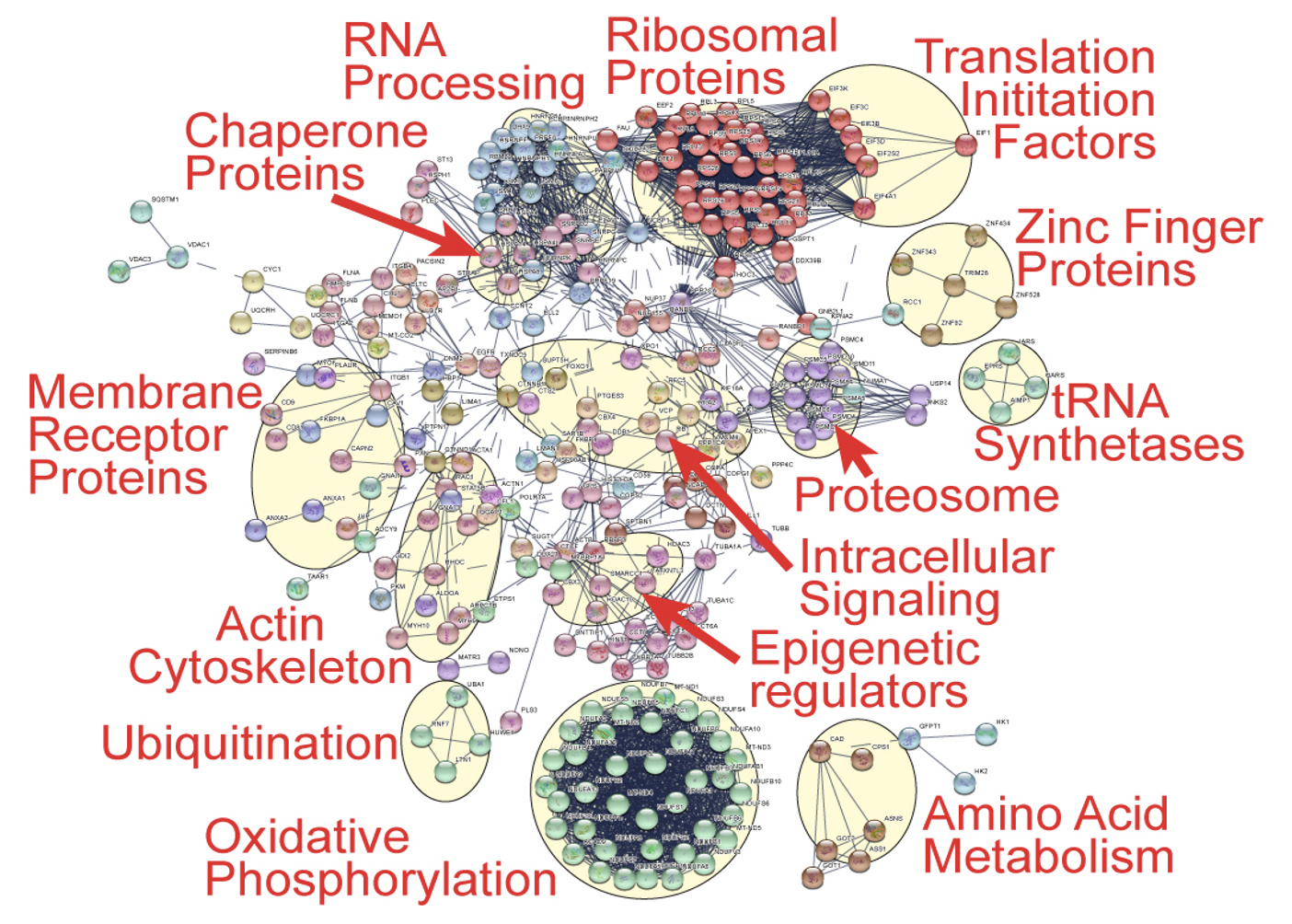
Potential histidine methylome
A protein interaction network describing the functional relationships among >200 identified histidine methylated proteins by ms. General biological categories are highlighted and labeled.